Dystrofie mięśniowe
| dystrophia musculorum progressiva | |
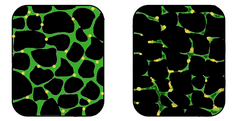 Dystrofia mięśniowa to zaburzenie genetyczne, w którym tkanka mięśniowa zanika i traci funkcje. W mięśniu dotkniętym chorobą (po prawej) tkanka uległa dezorganizacji, a stężenie dystrofiny (kolor zielony), ważnego białka w normalnym funkcjonowaniu mięśni, zostało znacznie zmniejszone. | |
| Klasyfikacje | |
| ICD-10 | G71.0 |
|---|---|
Dystrofie mięśniowe (zanik mięśni) – choroby mięśni objawiające się zmianami patologicznymi we włóknach mięśniowych i tkance łącznej. Są to choroby dziedziczne[1].
W chorobie tej dochodzi do zwyrodnień mięśni poprzecznie prążkowanych. Zanik mięśni jest przeważnie obustronny, a najczęściej mięśnie ksobne (tułowia) ulegają zwyrodnieniu. Nie stwierdza się zaburzeń czucia. Zmiany histopatologiczne polegają na powiększeniu grupy mięśniowej, podłużnym rozszczepieniu, zatarciu poprzecznego prążkowania, zeszkliwieniu i zaniku. Rozwija się tkanka łączna i tłuszczowa[2]. Zmiany w komórkach mięśniowych (miocytach) polegają na zaniku lub nieprawidłowej budowie białek – dystrofiny i utrofiny, których funkcją jest przymocowywanie miofibrylii do sarkolemmy.
Objawy i symptomy
- postępująca utrata umięśnienia
- zachwiana równowaga
- opadające powieki
- atrofia
- skolioza
- niezdolność do chodzenia
- częste upadanie
- kaczkowaty chód
- deformacja łydek
- ograniczony zakres ruchów
- trudności oddechowe
- skrócenie stawów
- kardiomiopatie
- zaburzenia rytmu serca
Podział dystrofii mięśniowych
Wyodrębniono trzy grupy chorób[1]:
- dystrofinopatie (postać Duchenne’a, Beckera i pośrednia, a także izolowana kardiomiopatia rozstrzeniowa sprzężona z chromosomem X, izolowana miopatia mięśni czworogłowych) – zależą od różnych mutacji tego samego genu odpowiedzialnego za brak lub ubytek białka komórki mięśniowej – dystrofiny[1];
- nukleopatie (dystrofie Emery-Dreifussa I i II, laminopatie, dystrofia miotoniczna oraz dystrofia oczno-gardzielowa) – wiążą się z defektem struktury białek błony jądra komórkowego[3];
- niejednorodną klinicznie i genetycznie grupa schorzeń mięśni, w których stwierdza się brak lub deficyt wielu białek lub kompleksów białkowych, pełniących ważne funkcje w komórce mięśniowej[1] – dystrofie obręczowo-kończynowe (LGMD, limb-girdle muscular dystrophies), dystrofia twarzowo-łopatkowo-ramieniowa.
Najczęstsze typy dystrofii
Dystrofia mięśniowa postępująca typu Duchenne’a (DMD)
Postać ta jest najczęstsza, chorują na nią osoby płci męskiej. Objawami w pierwszych latach życia jest upośledzenie ruchów, trudności w chodzeniu, w szczególności podczas wchodzenia po schodach oraz pionizacji z pozycji siedzącej. Z biegiem czasu pojawiają się zaniki mięśniowe. Wskutek zaniku mięśni powstają przykurcze mięśni, utrudniony chód (chód kaczkowaty), a także podczas wstawania z pozycji leżącej osoba wstaje na czworaka, prostując kończyny dolne, podpierając się kończynami górnymi. W badaniu radiologicznym wykrywa się w kościach długich zwężenie średnicy trzonów i jam szpikowych. Choroba rozwija się w ciągu od pięciu do piętnastu lat, podczas tego czasu następuje rozległy zanik oraz niedowład mięśni, który uniemożliwia chodzenie[1].
Dystrofia miotoniczna
Głównym objawem jest miotonia, czyli utrudnione lub opóźnione rozluźnienie mięśnia po skurczu, zanik i osłabienie mięśni oraz zaćma. Jest to choroba wielonarządowa, dolegliwości mogą dotyczyć także serca, układu pokarmowego i ośrodkowego układu nerwowego, mogą występować również zaburzenia hormonalne[4].
Postać twarzowo-łopatkowo-ramienna
W przypadkach typowych wyraża się osłabieniem mięśni twarzy i postępującym osłabieniem mięśni stabilizujących łopatki i zginaczy grzbietowych stóp, ale częste są także postacie nietypowe[5].
Dystrofia mięśniowa postępująca typu Beckera (DMB)
Osłabienie mięśni występuje później niż w dystrofii typu Duchenne’a, a przebieg choroby jest powolniejszy i łagodniejszy. Pierwszym objawy choroby pojawiają się między 1. a 4. dekadą życia, a także później. Średni wiek zachorowania to 12. rok życia. Pacjenci z tego typu dystrofią zachowują zdolność poruszania się do 4. dekady życia lub dłużej[1].
Dystrofie obręczowo-kończynowe (LGMD, limb-girdle muscular dystrophies)
Jest to niejednorodna klinicznie i genetycznie grupa schorzeń mięśni. Zidentyfikowano kilkanaście wariantów LGMD, a których każdy jest wynikiem mutacji innego genu. Pierwsze objawy pojawiają się przed 20. rokiem życia, niekiedy rozwój choroby następuje w znacznie późniejszym wieku. Początkowo występuje symetryczne osłabienie i zanik mięśni kończyn; wcześnie dochodzi do zniesienia odruchów kolanowych, pojawia się pogłębienie lordozy lędźwiowej, odstawanie łopatek i przykurcze ścięgien Achillesa[5].
Zobacz też
Przypisy
- ↑ a b c d e f ElżbietaE. Szmidt-Sałkowska ElżbietaE., MałgorzataM. Dorobek MałgorzataM., Nowe poglądy na patogenezę dystrofii mięśniowych postępujących (DMP): dystrofinopatii, nukleopatii, dystrofii obręczowo-kończynowych i dystrofii twarzowo-łopatkowo-ramiennej, „Polski Przegląd Neurologiczny”, tom 2 nr 3, Via Medica, ISSN 1734-5251 [dostęp 2021-06-21] .
- ↑ Zrozumieć chorobę [online], Fundacja Parent Project [dostęp 2021-06-21] .
- ↑ MałgorzataM. Dorobek MałgorzataM., ElżbietaE. Szmidt-Sałkowska ElżbietaE., Nukleopatie: emerynopatie i laminopatie – niejednorodność obrazu klinicznego, „Polski Przegląd Neurologiczny”, ISSN 1734-5251 [dostęp 2021-06-21] .
- ↑ AnnaA. Łusakowska AnnaA., AnnaA. Sułek-Piątkowska AnnaA., Dystrofia miotoniczna – nowe spojrzenie na znaną chorobę, „Neurologia i Neurochirurgia Polska”, tom 44 nr 3, Via medica, 2010, s. 264–276, ISSN 0028-3843 [dostęp 2021-06-21] .
- ↑ a b MałgorzataM. Dorobek MałgorzataM., ElżbietaE. Szmidt-Sałkowska ElżbietaE., Heterogenność genetyczna dystrofii obręczowo-kończynowych i niejednorodność kliniczna dystrofii twarzowo-łopatkowo-ramieniowej, „Polski Przegląd Neurologiczny”, tom 2 nr 3, Via Medica, 2006, ISSN 1734-5251 [dostęp 2021-06-21] .
 Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.
Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.
Kontrola autorytatywna (klasa chorób):
- Britannica: science/myotonic-dystrophy, science/muscular-dystrophy
- DSDE: muskelsvind











